sub function_name {
statement;
:
:
}
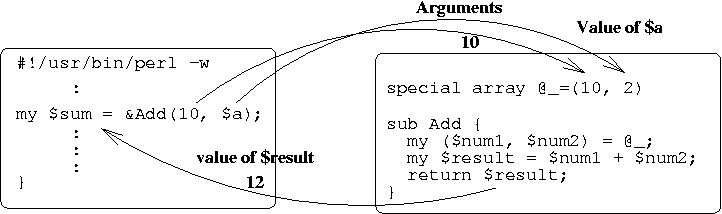
#!/usr/bin/perl -w
my $a = 2;
my $sum = &Add(10, $a);
print "10 + $a is $sum\n";
exit;
sub Add {
my ($num1, $num2) = @_;
my $result = $num1 + $num2;
return $result;
}
&function_name(argument1, argemuent2, ...);
or
function_name(argument1, argemuent2, ...);
&), indicating the subroutine
(c.f. $ indicates scalar variable), is optional (I usually
don't use it).
func1();
The returned value can be assigned to a variable.
But returning values is optional; some subroutines do not require a returning value.
#!/usr/bin/perl -w
my $seq = "ATGCAA";
my $numA = CntBase($seq, "A");
print "Num of Base A $numA\n";
exit;
sub CntBase {
my $seq = $_[0];
my $base = $_[1];
my @baseArray = split (//, $seq);
my $cntr = 0;
foreach my $b (@baseArray) {
if ($b eq $base) {
$cntr++;
}
}
return $cntr;
}
my $seq = $_[0]; my $base = $_[1];
Another method to receive the arguments:
my $seq = shift; my $base = shift;
The first element of @array gets assigned to $element, and
the 1st element gets removed from @array.
my @a = 10..13; # @a = (10, 11, 12, 13)
my $element = shift(@a); # $element = 10
# @a = (11,12,13)
If no argument is given to shift, it operates on the default array @_. That's why my $seq = shift; works.
sub BaseComposition {
my $seq = shift;
my @baseArray = split (//, $seq);
my %cntResult = (
"A" => 0,
"T" => 0,
"G" => 0,
"C" => 0
);
foreach my $b (@baseArray) {
$cntResult{$b}++;
}
return %cntResult;
}
$seqString = join ('', @baseArr); can be used to convert an
array to a character string.
my $var = "value";
my ($a, $b, $c, $d) = (1,2,3,4);
Don't forget ( ). The following is wrong:
my $a, $b, $c, $d; # only $a is private.
$globalVar = "Everyone knows me.";Or if you want to specify global, use our
our $globalVar = "Everyone knows me.";
$x and $y.
$x = "global 1";
$y = "global 2";
&func();
print "\noutside\n";
print "x = $x, y = $y\n";
exit;
sub func {
my $x = "local";
print "inside\n";
print "x = $x, y = $y\n";
$y = "global modified";
}
{ }.
#!/usr/bin/perl
$var1 = 1;
# just a block
{
my $var1 = 2;
print "IN: $var1\n";
}
print "OUT: $var1\n";
my $sum;
for(my $i=1; $i < 10; $i++) {
# a block for for loop.
$sum = $sum + $i;
my $wrongSum = $wrongSum + $i;
print "i=$i, sum=$sum, wrongSum=$wrongSum\n";
}
Note that I did not use /usr/bin/perl -w. What happens if
you use -w?
FASTA is a very simple format. There is 1 line which starts with ``>''. This line contains the name (identification) of this sequence. Then the sequence data come in several lines after the sequence name. The next sample start when you encounter the next ``>''.
> Camel-Prion GCTGACACCCTCTTTATTTTGC AGATAAATCATCATG > Camel-immunoglobulin GGACACGGCCGTTTATTACTGC GCACGTCGTACTATAGTGGTGT GTACTCGTACACC : :
You run the program like this:
./cntBase.pl camel.fasta
and the expected output is something like
> Camel-Prion 120 132 111 122
> Camel-immunoglobulin 24 32 11 22
The four numbers correspond to counts of bases (A, T, G, C) in each sequence.